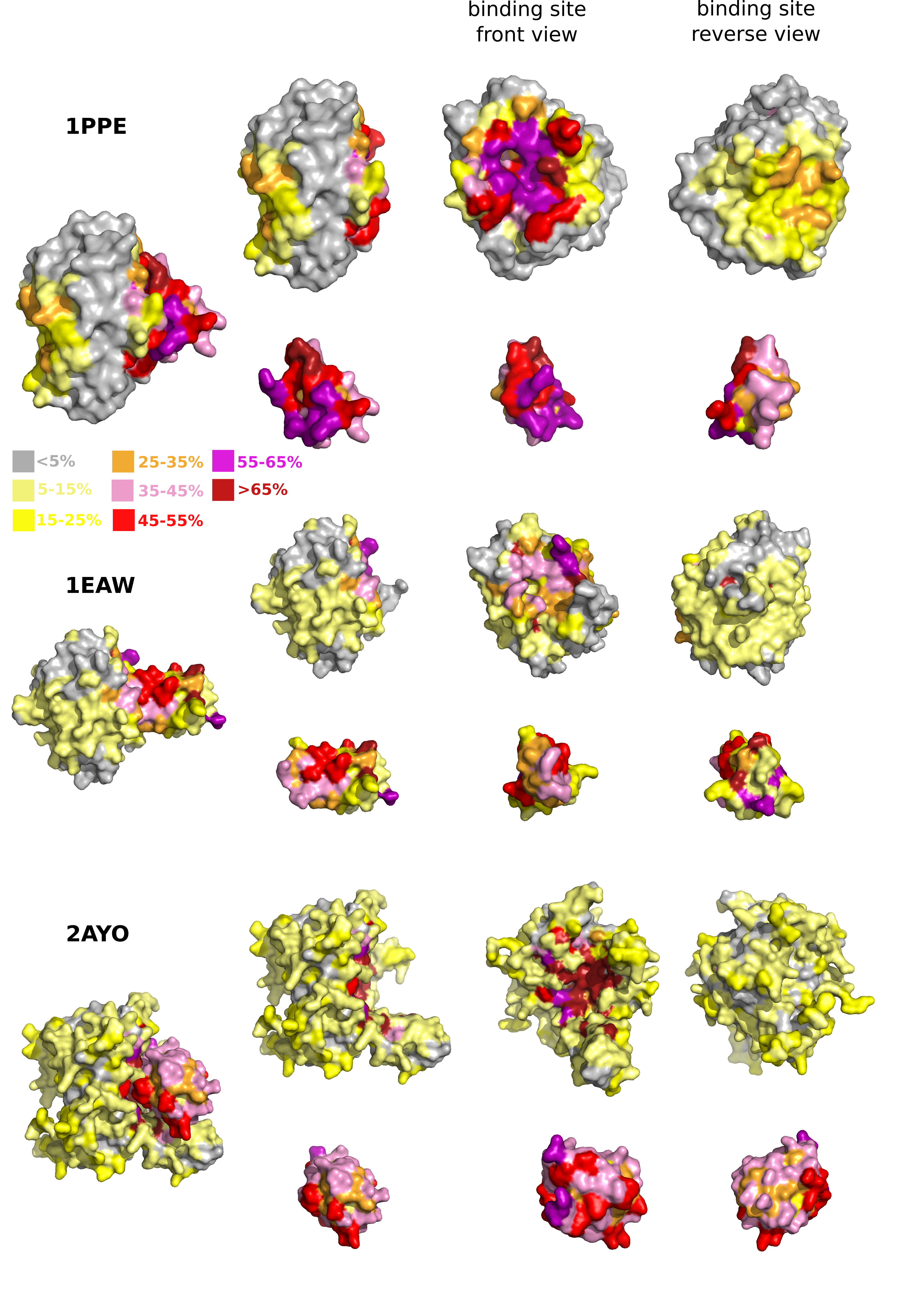
Complete Cross-Docking of PPDBv4
Protein social behavior makes a stronger signal for partner identification than surface geometry
Overview:
Cells are interactive living systems where proteins movements, interactions and regulation are substantially free from centralized management. How protein physico-chemical and geometrical properties determine who interact with whom remains far from fully understood. We show that characterizing how a protein behaves with many potential interactors in a complete cross-docking study leads to a sharp identification of its cellular/true/native partner(s). We define a sociability index, or S-index, reflecting whether a protein likes or not to pair with other proteins. Formally, we propose a suitable normalization function that accounts for protein sociability and we combine it with a simple interface-based (ranking) score to discriminate partners from non-interactors. We show that sociability is an important factor and that the normalization permits to reach a much higher discriminative power than shape complementarity docking scores. The social effect is also observed with more sophisticated docking algorithms. Docking conformations are evaluated using experimental binding sites. These latter approximate in the best possible way binding sites predictions, which have reached high accuracy in recent years. This makes our analysis helpful for a global understanding of partner identification and for suggesting discriminating strategies. These results contradict previous findings claiming the partner identification problem being solvable solely with geometrical docking.
Experiment:
We performed Complete Cross-Docking (CC-D) of the Protein Protein Docking Benchmark version 4.0 (PPDBv4, 176 complexes). Given a protein pair from the dataset, 2 docking calculations were performed where the 2 proteins alternatively played the role of the receptor and that of the ligand. In total, 447~040 docking calculations were realized. Docking was performed with HEX v6.3 using the shape complementarity scoring function. The precision of the molecular representation was defined from 18 and 25 expansion orders for the initial and final search steps. To avoid bias coming from the input PDB structures, the receptor and ligand models were positioned at a distance of 100 Angstroms from one another prior to docking. Moreover, five starting positions were defined using HEX Macro-Sampling module and were used to generate initial docking orientations for the ligand over the receptor and to derive appropriate local coordinate frames. In all docking calculations, we used the unbound conformations of the proteins from PPDBv4.
Download:
We provide for each protein the interaction propensities (IP) of its residues. The IP value of a residue is computed as the number of times it is present in a docked interface divivded by the number of retained conformations in the docking ensemble. Here, we retained 2000 conformations. The docked interfaces are defined by the sets of residues that display a change of at least 10% decrease in accessible surface area compared to the unbound protein. Te IP values are available here.
You can unpack the archive through the command
tar -zxf IPval.tgz
Contacts:
For questions, comments or suggestions feel free to contact Alessandra Carbone or Elodie Laine.
Reference:
If you are using our data, please cite:
- E. Laine and A. Carbone. (2017) Protein social behavior makes a stronger signal for partner identification than surface geometry. Proteins. 85:137-154.
Last Update September 2016

