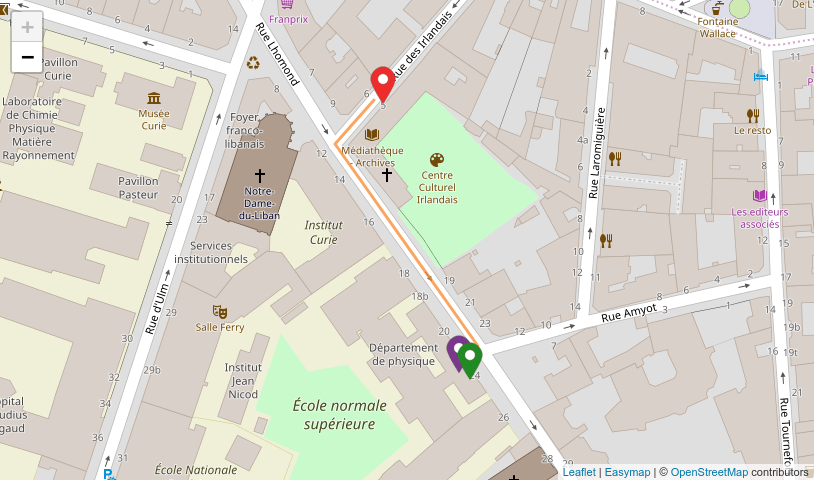
Friday 10th November 2017 Centre Culturel Irlandais @ 9am Salle Michel Guillaume - 5 rue des Irlandais, 75005 Paris
No fees. No pressure. All fun.
The Paris Biological Physics Community Day (PBPCD 2017) is a conference organized by young researchers of biological physics in the Paris area. We aim to bring together enthusiastic researchers in biophysics in the Paris area to create an opportunity for meeting and sharing knowledge.
The meeting is intended for researchers working in diverse areas of biophysics. It is going to be a day of conviviality and scientific enthusiasm, we envision to have a dynamic and informal atmosphere. In the program the talks of the invited speakers are interleaved with short presentations by young investigators.
The event is organized on behalf of GDRI (Groupement De Recherche International "Evolution, Regulation and Signaling") which also provides the funding.
No registration and no fees: lunch, coffee breaks and closing cocktail included!! Just come at the Centre Culturel Irlandais.
For any questions, contact us on the event page:
Keynote Speakers
Agnese SeminaraLaboratoire de Physique de la matière condensée, Université de Nice, FR
Christophe DanelonTU Delft/Department of Bionanoscience, NL
Sander TansAMOLF,Amsterdam, NL
Peter SwainSynthetic and Systems Biology, University of Edinburgh, UK
Program
9h00 - 9h30
Welcome
9h30 - 11h00
Christophe Danelon
Building a synthetic minimal cell
Gizem Ozbaykal
Mechanics of bending and cell shape recovery in Escherichia coli
Yanan Liu
The dynamics of flexible filaments in shear flow
11h00 - 11h30
Coffee Break
11h30 - 13h00
Agnese Seminara
Olfactory based navigation in mice
Min-Yeong Kang
A comprehensive model of gas exchange in the human lung
Pierre Illien
Exothermicity is not a necessary condition for enhanced diffusion of enzymes
Agnese SeminaraLaboratoire de Physique de la matière condensée, Université de Nice, FR
Olfactory based navigation in mice
Olfaction is one of the most obscure and fascinating sensory systems. Living organisms evolved to understand turbulence much better than us: a moth exposed to artificially smooth ("simple") odors, gets confused and is unable to navigate. Whereas moths in their natural environment use weak fluctuating turbulent odor signals to reach their female form hundreds of meters away. In this talk I will discuss olfactory navigation in mice, focusing on their ability to shift between multiple decision-making strategies, to strike a balance between flexibility and efficiency. I will first discuss the computational challenges involved in tracking a turbulent odor to its source. I will show that the signal bears information regarding the location of the source, and that this information is easy to extract if the animal is close to the source. Our behvioral experiments in an open arena show that mice are able to navigate to odor sources using naturally fluctuating airborne odor cues. When mice had limited prior experience of source locations, their search behavior was consistent with a gradient descent algorithm that utilized directional cues in the plume to navigate to the odor source. With increased experience, mice shifted their strategy from this flexible, sensory-driven search behavior to a more efficient and stereotyped foraging approach that varied little in response to odor plumes. This study demonstrates that mice use prior knowledge to adaptively balance flexibility and efficiency during complex behavior guided by dynamic natural stimuli.
Christophe DanelonTU Delft/Department of Bionanoscience, NL
Building a synthetic minimal cell
Building a molecular entity capable of self-reproduction and evolution is a formidable challenge in biology and evokes fascination among scientists and beyond. We are using a bottom-up synthetic biology approach to the construction of a minimal cell. Its chassis entails the expression of a minimal DNA genome inside self-assembled lipid vesicles. I will first describe the core architecture of our gene-based minimal cell. Then, I will present our results toward the functional assembly of three biological modules: DNA replication, compartment growth through lipid biosynthesis and vesicle division. I will finally discuss the remaining scientific challenges to the creation of synthetic life and our vision to tackle them.
Sander TansAMOLF,Amsterdam,NL
Single cell dynamics from bacterial growth to organoid infection
We use time-lapse microscopy to measure the dynamics of individual cells focusing on a number of diferent questions. Examples include the relation between 1fuctuations in the expression of catabolically active enzymes and cellular growth how cells control their size in the presence of external and internal perturbations and a surprising observation of motility in epithelial cells that is triggered by viral infection.
Peter SwainSynthetic and Systems Biology, University of Edinburgh, UK
An intracellular organization of extracellular information
Although cells respond specifically to environments, how environmental identity is encoded intracellularly is not understood. Here I will discuss this organization of information in budding yeast by estimating the mutual information between environmental transitions and the dynamics of nuclear translocation for ten transcription factors. Information is transduced through two channels - generalists and specialists - and transcription factors report differently and collectively can provide more information than predicted by noise-averaging. Changes in nuclear localization of multiple transcription factors thus constitute a precise, multi-dimensional internal representation of complex environments.
Short talks:
Alexandre DeloupyLaboratoire Jean Perrin, UPMC
Stochastic gene expression in bacillus subtilis
Stochastic gene expression in Bacillus subtilis Alexandre Deloupy, Lydia Robert, Vincent Sauveplane A population of genetically identical individuals sharing the same environment exhibits some residual diversity, referred to as phenotypic variability. This diversity can be the result of the stochastic nature of gene expression. We aim to precisely quantify the relative influence of transcription and translation on prokaryotic gene expression stochasticity, especially the effect of promoters, Transcription start Sites (TSS) and RBS. Cell to cell heterogeneity is measured in terms of noise strength. A stochastic model proposed by Thattai and van Oudenaarden in 2001 [1] predicts that noise strength varies linearly with translational efficiency but does not depend on transcriptional one. This prediction was shown to be compatible with data [2] on a limited number of strains and conditions but has never been fully tested on a large collection of strains with different transcription and translation efficiencies. We use such a collection of 49 strains of the bacterium Bacillus subtilis where GFP is expressed under the control of different Promoters, TSS and RBS. For each strain, cell-to-cell heterogeneity is investigated by quantifying fluorescence signal at the single cell level, based on flow cytometry techniques and epifluorescence microscopy. Using the precise quantification of expression noise in all strains we aim at improving our understanding of the effect of the different genetic sequences controlling protein expression, in particular promoters and RBS, thus further testing the prediction in [1], as well as the TSS, which can affect both transcription initiation and mRNA stability and folding. [1] : Thattai M. and van Oudenaarden A., (2001) Intrinsic noise in gene regulatory networks. PNAS. 98, 8614-8619. [2] : Ozbudak EM. et al., (2002) Regulation of noise in the expression of a single gene. Nature genetics. 31, 69-73.
Carmina Angelica Perez RomeroInstitut Curie and McMaster University
Studying transcription dynamics in live fly embryos
In embryos, cell differentiation occurs via the formation of spatial gradients of molecules called morphogens, which control the expression of a number of target genes determining cell identity. A common model system to study morphogens is the Bicoid gradient, which determines anterio-posterior (AP) patterning in Drosophila melanogaster, or fruit fly. We are especially interested in understanding how a noisy morphogen input can give a precise output of its target, and accomplish robustness during embryonic development. Here, we aim to apply novel methods in both fly genetics (to label the nascent mRNA of target genes) and fluorescence imaging (to detect the fluctuations in signal caused by the periodic creation of new mRNA at transcription sites) in order to measure the rate of transcription of the Bicoid target gene, hunchback, in each nucleus along the AP axis. Systematic measurements will allow us to determine which factors influence this transcription rate (e.g. morphogen diffusion rate, morphogen/target concentration, and polymerase activity at the target promoter), especially in the border region where there is a switch between expression and no expression. Given the rapidity of establishment of a precise transcriptional response, our hypothesis is that this response at the border relies on a memorization process, allowing nuclei to recall Bicoid concentration from one cycle to the next, by keeping trackof the Hunchback promoter transcriptional status across mitosis. Future experiments aiming to challenge this hypothesis, using novel methods in live embryonic imaging are discussed
Pierre IllienESPCI
Exothermicity is not a necessary condition for enhanced diffusion of enzymes
Recent experiments have revealed that the diffusivity of exothermic and fast enzymes is enhanced when they are catalytically active, and different physical mechanisms have been explored and quantified to account for this observation. We perform measurements on the endothermic and relatively slow enzyme aldolase, which also shows substrate-induced enhanced diffusion. We propose a new physical paradigm, which reveals that the diffusion coefficient of a model enzyme hydrodynamically coupled to its environment increases significantly when undergoing changes in conformational fluctuations in a substrate concentration dependent manner, and is independent of the overall turnover rate of the underlying enzymatic reaction. Our results show that substrate-induced enhanced diffusion of enzyme molecules can be explained within an equilibrium picture and that the exothermicity of the catalyzed reaction is not a necessary condition for the observation of this phenomenon. References: Exothermicity is not a necessary condition for enhanced diffusion of enzymes P. Illien, X. Zhao, K. K. Dey, P. J. Butler, A. Sen, R. Golestanian Nano Lett. 17, 4415 (2017) Diffusion of an enzyme: the role of fluctuation-induced hydrodynamic coupling P. Illien, T. Adeleke-Larodo, R. Golestanian to appear in Europhys. Lett., arXiv:1611.02580.
Gizem OzbaykalInstitut Pasteur
Mechanics of bending and cell shape recovery in Escherichia coli
Bacterial cell shape is determined by the peptidoglycan cell wall. For rod-shaped E. coli cells, mechanical bending experiments have shown that cells adapt to long-term mechanical bending forces by assuming a bent cell shape, and bent cells straighten over time if the force is removed. However, the physical mechanisms underlying these processes are not known. It was previously shown that the bacterial actin MreB localizes specifically to the inner sides of bent cells, and it was suggested that MreB localization is responsible for cell straightening. In this study, we monitor cell shape and MreB localization during cell bending and cell straightening using microchambers. We show that MreB localization is indeed increased at the inner sides of both bent and straightening cells in a curvature dependent manner. Yet, the degree of differential localization is not sufficient to explain the observed rate of straightening. Furthermore, MreB localization can also not be responsible for cell bending, as MreB is predominantly found at the inner side of the bent cell, where the cell wall elongates less. Instead, a coarse-grained model developed by our collaborators, Ariel Amir and Felix Wong, can reconcile both bending and straightening processes by coupling processive cell-wall synthesis to the differential degree of cell-wall stretching on the different sides of the cell.
Min-Yeong KangLaboratoire de la Physique de la Matière Condensée (PMC), Ecole Polytechnique
A comprehensive model of gas exchange in the human lung
A comprehensive model of gas exchange in the human lung. Despite its importance, the gas exchange function of the lung has not been comprehensively understood yet. The current understanding is either based on very rough approximation, using an analogy of single balloon supplied air by a single tube, neglecting all the structural complexity or focused on some part of the lung. Here we propose a first comprehensive model of the gas exchange. Meaning that, for given respiratory parameters it enables a quantitative prediction of the gas exchange in terms of the oxygen capture and the blood saturation. This was possible by constructing a set of equations for gas convection-diffusion-permeation in the airways and oxygen-hemoglobin saturation in the pulmonary capillaries for a pulmonary acinus, a unit of gas exchange in the lung. This equation set relates the condition of air and blood entering into the lung with the resulting gas exchange. Predictions of O2 capture in some typical respiration conditions will be presented. And its clinical application to obtain parameters that is difficult to measure such as cardiac output will be also introduced briefly.
Zhanna SantybayevaCBS, CNRS UMR 5048
Photonic Force Microscopy: topographical imaging at femto-Newton forces
The biggest limitation of the well-acclaimed technique for viscoelastic and topographical measurements, the Atomic Force Microscopy (AFM), is intrinsic and is hidden in its design, more precisely, in its cantilever. We present an analogous system under construction, a Photonic Force Microscope (PFM), which is based on optical trapping and overcomes this limitation. It permits to reconstruct the topography of soft biological and non-biological samples with simultaneous local rheology [1, 2]. We use the back focal plane interferometry for detection of the positions of a probe in the optical trapping volume with nanometric precision. Compared to the AFM, the absence of a rigid cantilever is an advantage, and the stiffness of the trap and thus the applied force can be adjusted dynamically. For comparison, the typical forces in the AFM are in the range of 0.1-100 pN [3] and can lead to displacement or even destruction of super-soft biological samples such as lipid bilayers and living cells [4]. Moreover, the thermal noise of the PFM is beneficial with respect to that in the AFM and is used as a measure of local viscosities. We both produce and use probes of different configurations, e.g. micrometric cones and cylinders with small protrusions of quartz for potentially better axial resolution, and more classical polysterene microspheres. By scanning the sample across the fixed in the trap probe and using the phase correction method [5], we are able to obtain a 3D topography of the sample. References [1] E.-L. Florin, A. Pralle, J.K.H. Hörber, E.H.K. Stelzer, J. Struct. Biol. 1997 119, 202-211 [2] F. Juenger, F. Kohler, A. Meinel, T. Meyer, R. Nitschke, B. Ehrhard, A. Rohrbach, Biophysical Journal, 2015, 109, 869-882 [3] K. C. Neuman, A. Nagy, Nature Methods, 2008, 5 (6), 491-505 [4] A. Pralle, E.-L. Florin, E.H.K. Stelzer, J.K.H. Hörber, Single Molecules, 2000, 1 (2), 129-133 [5] P. C. Seitz, E.H.K. Stelzer, A. Rohrbach, Applied Optics, 2006, 45 (28), 7309-7315
Yanan LiuPMMH - ESPCI
The dynamics of flexible filaments in shear flow
The rheological properties of complex fluids made of particles in a suspending fluid depend on the behavior of the microscopic particles under flow [1]. A first step to understand this link is to investigate the individual particle dynamics in different flow geometries. A rigid rod will perform so-called Jeffery orbits, however, when the rigid rod becomes flexible and Brownian, the behavior in terms of deformation and rotation still needs to be fully understood [2]. We chose here to address this situation by studying the behavior of flexible polymers experimentally and numerically. In experiments, we use actin filament and combine fluorescent labeling techniques, microfluidic devices and an automated stage to carry out controlled systematical experiments and statistical analysis. In simulations, we model these inextensible filaments using Euler-Bernoulli beam elasticity and use non-local slender body theory (SBT) in the presence of Brownian fluctuations [3] to probe their dynamics. By increasing either the shear rate or the length of the filaments, we subsequently observe tumbling, buckling, and bending under flow.The evolution and transition of these typical dynamics are governed by the elasto-viscous $8\pi\mu\dot{\gamma}L^4/l_pk_BT$ number, comparing viscous forces to elastic restoring forces. There is a good agreement between experiments and simulations. Reference [1] Becker, Leif E., and Michael J. Shelley. "Instability of elastic filaments in shear flow yields first-normal-stress differences." Physical Review Letters 87.19 (2001): 198301. [2] Harasim, Markus, et al. "Direct observation of the dynamics of semiflexible polymers in shear flow." Physical review letters 110.10 (2013): 108302. [3] Manikantan, Harishankar, and David Saintillan. "Buckling transition of a semiflexible filament in extensional flow." Physical Review E 92.4 (2015): 041002.
Francesca RizzatoLPT & LPS - ENS
Inferring protein mutational landscapes from sequence data
Since the nineties, a large variety of statistical tools have been developed to extract, from genomic datasets, information which used to be attainable only through long and expensive experimental procedures. In the context of protein sequence data, one of these approaches is Direct Coupling Analysis (DCA) [Weigt et al, PNAS, 2009], a statistical inference method capable of effectively predicting protein three-dimensional structures from multiple sequence alignments (MSA) [Ekeberg et al., PRE, 2013] and more recently applied to the prediction of the effect of a mutation on protein fitness [Figliuzzi et al., MBE 2016 - Hopf et al., Nat. Biotech.,2017]. We present a modified approach of DCA that performs inference only on site-dependent subsets of the 20 amino acids [Coucke, Rizzato et al., in preparation] and we show its performances in predicting some protein fitness landscapes. This approach, by reducing the number of the parameters to be inferred, decreases the computational time of the inference process without loss of accuracy in the fitness predictive power.
Location
Talks & Coffee Breaks:
Centre Culturel Irlandais, Room Michel Guillaume 5 rue des Irlandais
Lunch:
École normale supérieure, Rooftop - Serre room 24 Rue Lhomond see map
Cocktail:
École normale supérieure, Rooftop - Serre room 24 Rue Lhomond see map

{kind=link}