|
|
|
|
The Île-de-France Region has defined its 9 new major areas of research and innovation for the next 5 years, in which €100M will be invested.
Among them:
► AI4IDF - Intelligence artificielle centrée sur l’humain en Île-de-France, lead by Inria - PIs: Isabelle Ryl
► BIOCONS - Bioconvergence pour la santé, lead by Université de Paris Cité - PIs: Amanda Sylva Brun and Ariel Lindner
The LCQB participates in both programs.
|
|
|
Towards predicting new mutations in SARS-CoV-2: in the very early stages of an emerging viral outbreak, can we predict new mutations in the proteins of the virus which might lead to its future variants? The "Statistical Genomics and Biological Physics" team proposes a computational approach based on a single viral genome infecting humans and pre-existing viral genomes infecting other species.
For more information see:
|
Gene duplication is a major source of functional diversification. However, the co-existence of two paralogues with similar biochemical properties but diverging functions can lead to potentially detrimental competition between the duplicates. This phenomenon has been named “paralogue interference”. In a recent article published in "Frontiers in cellular infection and microbiology, the "genetics networks" team of LCQB has addressed this question in the human pathogen Candida glabrata. They showed that evolution selected mutations which decreased competition between two, potentially interfering, transcription factors, thus allowing the emergence of the particular modes of regulation for respiration and iron homeostasis observed in extent Saccharomycetaceae species.
Link to the article
|
|
|
|
|
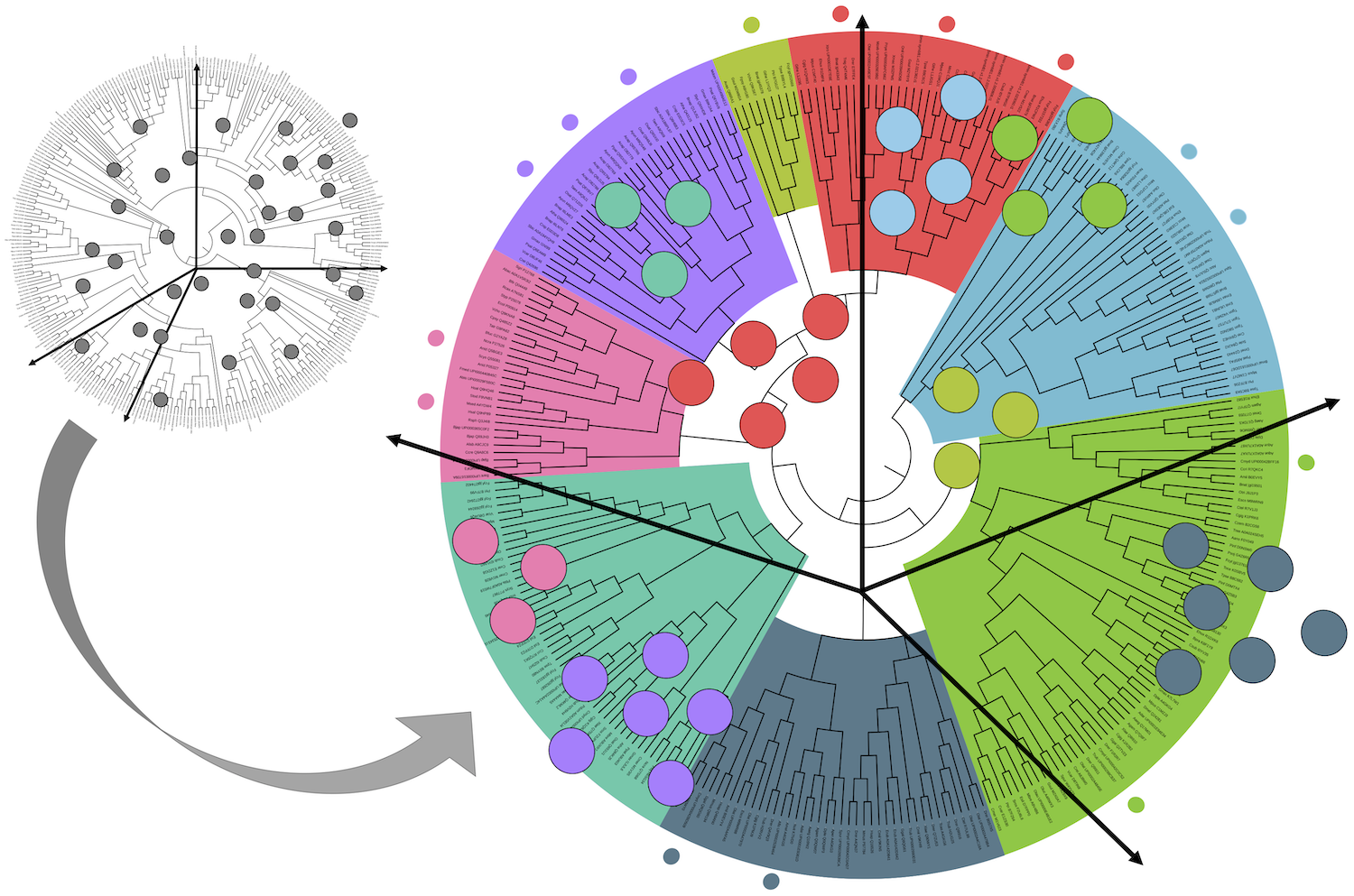
The analysis of the subtelomeres of a green alga, published in Nucleic Acids Research, is featured in this press release on the INSB website. The work, a collaboration between the LCQB ("Telomere and Genome Stability" and "Biology of Genomes" teams) and the IBPC, sheds light into the complexity and evolution of the subtelomeres, structures that are critical for genome integrity.
|
|
|